Isolation of DNA from bacteria
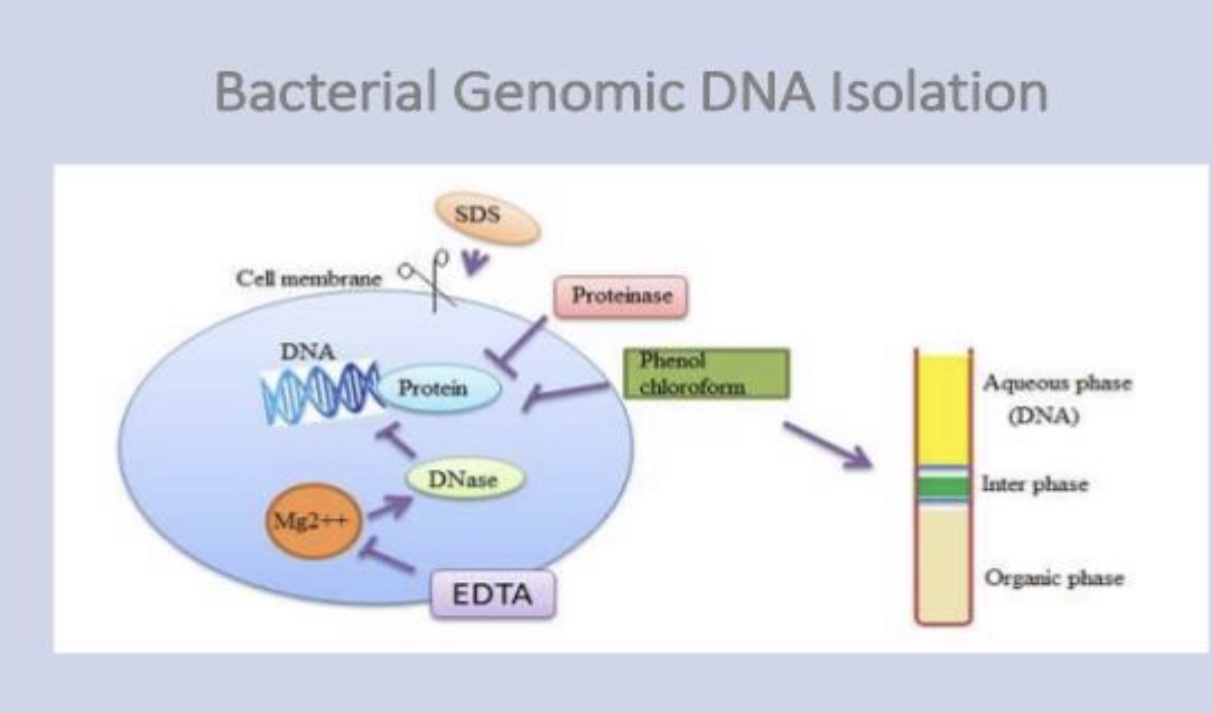

The isolation of DNA from bacteria is a relatively simple
process. The organism to be used should be grown in a favorable medium
at an optimal temperature and should be harvested in a late log to early
stationary phase for maximum yield. The genomic DNA isolation needs to
separate total DNA from RNA, protein, lipid, etc. Initially, the cell
membranes must be disrupted in order to release the DNA in the extraction
buffer. SDS (sodium dodecyl sulphate) is used to disrupt the cell membrane
RNA can be removed by treatment with ribonuclease (RNase) that has
been heat-treated to inactivate any DNase contaminants; RNase is
relatively stable to heat as a result of its disulphide bonds, which ensure
rapid renaturation of the molecule on cooling. The other major
contaminant, protein, is removed by shaking the solution gently with
water-saturated phenol, or with a phenol/chloroform mixture, either of
which will denature proteins but not nucleic acids
.
The deproteinised DNA
preparation is mixed with two volumes of absolute ethanol, and the DNA
allowed to precipitate out of solution in a freezer. After centrifugation, the
DNA pellet is redissolved in a buffer containing EDTA to inactivate any
DNases present. This solution can be stored at 4oC for at least a month.
DNA solutions can be stored frozen although repeated freezing and
thawing tends to damage long DNA molecules by shearing. The procedure
described above is suitable for total cellular DNA. If the DNA from a specific
organelle or viral particle is needed, it is best to isolate the organelle or
virus before extracting its DNA, since the recovery of a particular type of
DNA from a mixture is usually rather difficult. Where a high degree of purity
is required DNA may be subjected to density gradient ultracentrifugation
through caesium chloride which is particularly useful for the preparation of
plasmid DNA
- .Net Interview Questions And Answers
- All Interview Questions And Answers
- Android Common Tips Q and Ans
- AJAX Interview Questions And Answers
- Codeigniter Interview Questions And Answers
- Aptitude Interview Questions And Answers
- C Interview Questions And Answers
- CSS3 Interview Questions And Answers
- Data Structure Questions With Answers
- Database (DBMS) Questions With Answers
- Drupal Interview Questions And Answers
- Download Career Guide in doc file
- Dojo Interview Questions And Answers
- Design Pattern of all type
- EJB Interview Questions And Answers
- Header Function use in PHP and HTTP
- HR Interview Questions And Answers
- HTML5 Interview Questions And Answers
- Joomla Interview Questions And Answers
- Iphone Interview Questions And Answers
- MYSQL Interview Questions And Answers
- Java Interview Questions And Answers
- JQuery Interview Questions And Answers
- JSON Interview Questions And Answers
- JSP Interview Questions And Answers
- linux Commands and Interview Questions
- Moodle Tutorial for Developers
- Magento Common Tips Q and Ans
- Networking Hardware Questions with Answers
- Operating Systems Interview Questions
- OOPs Interview Questions and Answers
- PHP Interview Questions And Answers
- PHP Interview Questions 1500+
- PHP All Objective Questions Answers
- PHP Jobs for freshers and experienced
- Project Management Interview questions
- Regular Expressions Interview questions
- Spring Interview Questions Answers In Java
- Software Testing Interview Questions Answer
- Servlets Interview Questions And Answers
- Struts Interview Questions And Answers
- Threads Interview Questions And Answers
- US Jobs city Wise
- All Indian Company Name List
- Web Designing Interview Questions Answers
- XML Interview Questions Answers
- XML Interview Questions Answers
- .net Interview Questions And Answers
- Accountant Interview Questions And Answers
- Ado.net Interview Questions And Answers
- Adp Interview Questions And Answers
- Agile Methodology Interview Questions And Answers
- Android Interview Questions And Answers
- Apache Interview Questions And Answers
- Application Packaging Interview Questions And Answers
- Asp Interview Questions And Answers
- Backbone.js Interview Questions And Answers
- C Sharp Interview Questions And Answers
- C++ Interview Questions And Answers
- Cake Interview Questions And Answers
- Cakephp Interview Questions And Answers
- Can Protocol Interview Questions And Answers
- Catia V5 Interview Questions And Answers
- Ccna Interview Questions And Answers
- Checkpoint Firewall Interview Questions And Answers
- Control M Interview Questions And Answers
- Cpp Interview Questions And Answers
- Css Interview Questions And Answers
- DATA GRID Interview Questions And Answers
- Data Warehouse Interview Questions And Answers
- Data Structures Interview Questions And Answers
- Database Interview Questions And Answers
- Db2 Interview Questions And Answers
- Desktop Engineer Interview Questions And Answers
- Desktop Support Interview Questions And Answers
- DOJO Interview Questions And Answers
- Electrical Engineering Interview Questions And Answers
- Embedded Systems Interview Questions And Answers
- Hadoop Interview Questions And Answers
- Hibernate Interview Questions And Answers
- J2ee Interview Questions And Answers
- Javascript Interview Questions And Answers
- Joomla Interview Questions And Answers
- Jsp Interview Questions And Answers
- Less Interview Questions And Answers
- Linq Interview Questions And Answers
- Linux Interview Questions And Answers
- Matlab Interview Questions And Answers
- Mcitp Interview Questions And Answers
- Netbackup Interview Questions And Answers
- Node.js Interview Questions And Answers
- Oracle Interview Questions And Answers
- Perl Interview Questions And Answers
- Plsql Interview Questions And Answers
- Postgresql Interview Questions And Answers
- Python Interview Questions And Answers
- Qa Testing Interview Questions And Answers
- Qtp Interview Questions And Answers
- Sap Interview Questions And Answers
- Sass Interview Questions And Answers
- Selenium Interview Questions And Answers
- SEO Interview Questions And Answers
- Sharepoint Interview Questions And Answers
- Silverlight Interview Questions And Answers
- Sql Dba Interview Questions And Answers
- String Interview Questions And Answers
- Struts2 Interview Questions And Answers
- Stware Testing Interview Questions And Answers
- Swing Interview Questions And Answers
- Technical Support Interview Questions And Answers
- Telecom Billing Interview Questions And Answers
- Tomcat Interview Questions And Answers
- Troubleshooting Interview Questions And Answers
- Uml Interview Questions And Answers
- Us Visa Interview Questions And Answers
- Vb Interview Questions And Answers
- Visa Interview Questions And Answers
- Wcf Interview Questions And Answers
- Web Testing Interview Questions And Answers
- Windows Interview Questions And Answers
- Wordpress Interview Questions And Answers
- Wpf Interview Questions And Answers
- XQuery Interview Questions And Answers
- Yii Interview Questions And Answers
- Zend Framework 2 Interview Questions And Answers
- Zend Framework Interview Questions And Answers